Clustering (CLUSTER)
Overview
Cluster Module for Automated Chemical Space Exploration
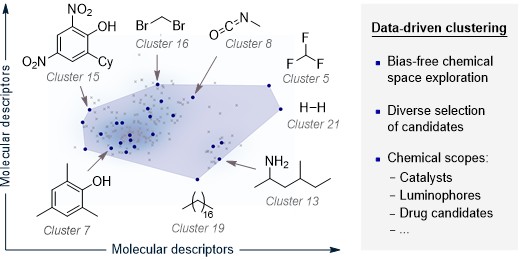
Clustering is an unsupervised learning technique used to partition high-dimensional chemical spaces into groups of structurally or functionally similar compounds, based on molecular descriptors. Within the ALMOS framework, the clustering module enables the systematic selection of an initial, diverse set of candidate molecules to seed active learning (AL) workflows. This approach reduces human bias in the selection process and ensures representative coverage of the chemical space. It is particularly suited for scenarios where defining the scope of a molecular library is critical, as it facilitates automated, scalable, and reproducible organization of chemical diversity.
How Cluster works
The current CLUSTER workflow in ALMOS is coverage-driven. Instead of asking the user to define a fixed number of clusters, the program:
builds a cleaned descriptor space from the input data,
evaluates several clustering strategies internally,
estimates a reasonable number of representative points automatically when
--n_pointsis not provided,and selects a diverse
batch = 0seed set for downstream active learning.
The default workflow is oriented toward representative selection, not manual cluster-count tuning.
Required Input
There are two main options:
The user can provide a CSV file containing molecular features (i.e., DFT properties, experimental measurements, etc.). In such cases, the '--name' option must be specified (e.g.
--name molecules).
cluster --input EXAMPLE.csv --name MOL_NAME
The user can also provide a CSV file containing the code_name and SMILES columns. In this case, ALMOS can automatically generate the molecular features from SMILES strings using the
--aqmeoption, implemented via the AQME package. This route is intended for users who do not already have a descriptor matrix.
cluster --input EXAMPLE.csv --name code_name --aqme
Main parameters
The most commonly used options in the current CLUSTER workflow are:
--input: input file for clustering. This is usually a CSV containing either descriptors or SMILES-based inputs to be processed with AQME.--name: identifier column used to track the selected molecules or conditions. This should contain unique labels for each row.--n_points: number of representative points to select. If omitted, ALMOS estimates a suitable selection size automatically.--y: optional target column used only for visual interpretation in the chemical-space viewer and related plots.--ignore: columns that should not be treated as descriptors. This is useful for names, textual metadata or bookkeeping columns.--aqme: enables descriptor generation from SMILES-based inputs before the clustering workflow starts.--evaluate: skips reselection and evaluates an already existingbatch = 0selection in the input file. This is useful when the user wants ALMOS to analyse a manual or external seed set instead of creating a new one.
Typical command-line patterns:
cluster --input EXAMPLE.csv --name MOL_NAME
cluster --input EXAMPLE.csv --name MOL_NAME --n_points 40
cluster --input EXAMPLE.csv --name MOL_NAME --ignore "[batch,SMILES]"
cluster --input EXAMPLE.csv --name code_name --aqme
cluster --input EXAMPLE.csv --name MOL_NAME --evaluate
CLUSTER Protocol in ALMOS
The user supplies a CSV file containing molecular descriptors, or a CSV/structure file that can be processed through
--aqme.ALMOS cleans the descriptor space and evaluates clustering quality internally.
The program selects a representative subset of points and writes them as
batch = 0in the output CSV.ALMOS generates a chemical-space viewer, coverage diagnostics and descriptor-based interpretation plots to help assess how representative the selected batch is.
If a dataset already contains a user-defined
batch = 0selection, the user can skip reselection and evaluate that selection directly with--evaluate.
Example
An example is available in Examples/Use of individual modules.